Input Files
Contents
- General
- Multiple Protein Sequence Alignment
- Gene Structure Files
- Mapping Sequence Names to Species Names
- Phylogenetic Tree Files
| General | Back to top |
GenePainter requires two types of input:
- A FASTA formatted multiple sequence alignment (MSA)
- A folder containing gene structures in YAML format as specified by WebScipio or in GFF v.3 format.
GenePainter combines information from the alignment with the gene structures. Therefore, the protein names from the MSA must match to the YAML or GFF filenames. Only those genes, which can be matched (i.e. protein name equals the YAML or GFF filename), will be analysed.
If gene structure information shall be plotted onto phylogenetic trees, GenePainter also requires either:
- NCBI taxonomy database dump
- User-provided text-file containing taxonomic lineages
| Multiple Protein Sequence Alignment | Back to top |
This file must be a multiple protein sequence alignment, in which all sequences are of same length. Protein sequences are matched with the gene structures on the basis of the FASTA header and file names, respectively. To this end, the FASTA header must be exactly like the corresponding YAML or GFF filename for each gene, which should be included in the analysis. For this reason, FASTA headers must not contain any blanks or special characters.
| Gene Structure Files | Back to top |
For the analysis, GenePainter needs gene structure information for each gene. This information must be stored either in the YAML format as generated by WebScipio or in GFF v.3 format. Moreover, all gene structures must be located in the same directory.
GFF
GFF3 formatted gene structures are parsed for "CDS" features. All "CDS" features are considered, which are linked via the attribute column (parent tag) to the first "mRNA" feature specified. If no mRNA feature is given in the GFF file, all CDS features are parsed. GenePainter uses column 1 (seqid), columns 4 and 5 (start and end of feature) and column 8 (phase). All other features and columns are ignored. GFF files generated by WebScipio can also be used as input, although they do not strictly follow GFF3 conventions.
YAML
The most convenient way to obtain YAML formatted files is to use the WebScipio web interface for gene reconstruction and to download the resulting YAML files. For automation of the YAML generation, several scriptable alternatives exist. First, WebScipio can be accessed by its web service API. This can be done within any software program. In the GenePainter package, scripts are included for querying WebScipio with genes belonging to a single species ( generate_yaml_for_species.rb ) or with genes belong to different species ( generate_yaml_for_multiple_species.rb ). Both scripts access WebScipio through the web service and store the resulting YAML files locally. A brief introduction to the usage of the web service can be found at the WebScipio homepage. Second, Scipio can be used locally, which requires further software (BLAT, Bioperl, YAML Perl module) and respective genome assembly files.
A list of all species available can be found at http://www.webscipio.org/webscipio/genome_list.
Usage of generate_yaml_for_species.rb
$ ruby tools/ generate_yaml_for_species.rb -s ‘Species name’ -i fasta_sequence.fas
Mandatory Arguments
| -s or --species <‘species_name‘> | Species encoding the specified protein(s). Species should be wrapped with "‘" to preserve spaces. | |
| -i or --input <file> | Path to fasta formatted protein sequence(s). Might be a multiple sequence alignment of sequences encoded by same species. | |
Optional Arguments
| -o or --outfile <file_name> | Name of the YAML output file. ONLY used if the fasta file contains only one sequence. Default: Use fasta header of the input protein sequence. | |
| -h or --help | Show help message. | |
Usage of generate_yaml_for_multiple_species.rb
$ ruby tools/ generate_yaml_for_multiple_species.rb -s example/fastaheaders2species.txt -i fasta_sequence.fas
Mandatory Arguments
| -s or --species-to-fasta <file> | Text based file mapping fasta header to species names. Mandatory line format: Fastaheader1[,Fastaheader2]:Species | |
| -i or --input <file> | Path to fasta formatted multiple sequence alignment. | |
Optional Arguments
| -h or --help | Show help message. |
YAML files will be named like the corresponding fasta headers. YAMLs are only generated for those sequences, for which a species is specified.
Structure of YAML files
Scipio and WebScipio store gene structure information in YAML format. This format comprises a collection of key – value pairs, an associative array. However, the accurate gene structure representation requires more keys than necessary for the alignment of the gene structures. Thus, GenePainter ignores some data included in the YAML files. Accordingly, these additional keys need not be included in manually reconstructed YAML files. A minimal working example YAML file is defined in Figure 1. An exhaustive description of all keys used by WebScipio can be found at the WebScipio homepage http://www.webscipio.org/help/scipio#description.

Figure 1 Excerpt from the YAML file describing HsCoro1A. All exon and intron descriptions but the very first and last ones have been omitted (marked in yellow). Blank lines were added to separate exon and intron descriptions. Additionally, green boxes highlight exons and blue boxes highlight introns. Only those key – value pairs, which are relevant for GenePainter are shown. The original YAML file is part of the test data included in the package.
The list of exons and introns ("matchings") must start with the keyword - matchings:. The order of keys describing the exons and introns is not important. Mandatory keys for successful incorporation in GenePainter are listed in the following tables:
| YAML keys | Description | type | "intron", "intron?", "exon", or "gap". "intron?" is used for uncertain introns (unusual splice patterns found) |
|---|---|
| nucl_start | Location in the query (in nucleotide coordinates). |
| seq | DNA sequence of the feature. |
| nucl_end (appears in exons only) | Location in the query (in nucleotide coordinates). |
| Mapping Sequence Names to Species Names | Back to top |
Only protein/gene sequence names that are linked to species names are considered for taxonomic computations or generation of YAML files, respectively. All other genes are omitted from these computations. Species names must match NCBI taxonomy, if YAML files are obtained via WebScipio and if the NCBI taxonomy tree is used for intron gain and loss plotting. The file containing the mapping of fasta headers and species names must be in a specific format (Figure 2): Only one species name per line. Gene and species names are separated by ":". If several genes correlate to the same species, their fasta headers might be given in one line. The list of fasta headers needs to be comma-separated or semicolon-separated . The species name must be enclosed by double quotes. Blanks immediately before and after ";" are permitted, and will be ignored by the program.
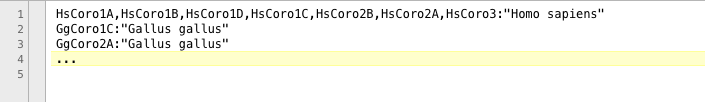
Figure 2 For taxonomy options as well as to generate YAML files, a mapping between genes described in MSA and corresponding species must be established. To this end, fasta headers (separated by ",") are mapped to species names. Fasta headers and species names are separated by ":".
| Phylogenetic Tree Files | Back to top |
To map intron gain and loss events onto phylogenetic trees, GenePainter needs to correlate the protein sequence/gene structure names and the species names as present in the NCBI taxonomy database or the user-provided list.
GenePainter can extract the taxonomic lineages from the NCBI taxonomy database dump, which can be obtained using the script download_taxdump.rb, or from a user-provided text-file containing taxonomic lineages (Figure 3). In this file, one lineage should be given per line. Taxa should be ordered from root to species and be separated from each other by semicolons. Optionally, a blank may follow the semicolon.

Figure 3 Excerpt of file providing a user-defined taxonomic lineage. For displaying purpose, parts of the lineage were omitted.




